
transformer
一、背景1. 序列到序列模型
1)语音辨识输入输出关系:输入声音信号(t帧特征向量),输出对应文字(n个字),t与n无固定比例关系。
典型案例:台语语音辨识系统(1500小时乡土剧训练数据)。
技术特点:直接端到端训练,跳过音标转换等中间步骤(”硬train一发”方法)。
2)机器翻译长度关系:输入句子长度n与输出句子长度n’由模型动态决定。
中英案例:“机器学习”(4字)→”machine learning”(2词),非固定1/2比例(可长可短)。
3)语音翻译(语音辨识复合机器翻译)特殊价值:适用于无文字语言(全球7000+语言中过半无文字系统)。
实现方式:直接建立语音到目标语言文字的映射,避免传统ASR+MT串联方案。
4)台语语音辨识应用案例
数据来源:YouTube乡土剧(台语音频+中文字幕对齐数据)。
技术挑战:背景⾳乐/噪声干扰(直接忽略处理);字幕与语音不完全对齐(仍可训练);跳过台罗拼音中介步骤(端到端训练);强行训练。
典型错误:倒装句处理困难(”我有帮厂长拜托” vs 正确应为”我拜托厂长”);语义理解偏差(”要生了吗” vs 正确答案”不会 ...

自注意力机制
自注意力机制自注意力(Self-Attention)机制是一种特殊的注意力机制,它允许模型在处理一个序列时,考虑到序列中每个元素与其他所有元素的关系。这种机制可以帮助模型更好地理解序列中的上下文信息,从而更准确地处理序列数据。 (序列数据是一种数据类型,其中的元素存在特定的顺序。每个元素都有特定的位置,且位置之间的顺序关系对于数据的含义和处理方式有重要影响)
在自注意力机制中,模型会计算序列中每个元素与其他所有元素的关联度(或称为权重)。这些权重反映了元素之间的相互关系,比如在语言模型中,它们可以反映词与词之间的语义关联度。(比如机器翻译)
1举例:以"the 2022 Beijing Winter Games"为例,当模型处理"Games"这个词时,通过自注意力机制,它可以考虑到"2022"、"Beijing"和"Winter"这些词的信息。这样,模型就能理解到"Games"在这里并不是指一般的"游戏",而是指特定的"比赛", ...
生成式对抗网络
生成式对抗网络(GAN)传统的对应的架构,生成架构
用network当作生成器,每一次随机生成对应的z,知道对应的分布是简单的,已知的均匀分布或者是正态分布,network的输出变成复杂的范围。
这样的网络就叫做生成器。
为什么要选择分布?训练资料是不断录制的影片,影片中小精灵的位置预测。传统的网络预测会让小精灵进行分裂,同样的转角会产生两种不同的结果,两种训练资料,进行两面讨好,就会同时向左向右转。让机器的输出是有几率的为其解决方法。加上一个分布包含向左转和向右转。
当我们的任务需要一点创造性时,同样的输入,多种不同的输出。
让人们进行画图的工作会产生不同的结果,比如画一个红色的眼镜的人。
采用聊天的方法,去生成对应的图片。
这样我们就要生成式对抗网络。
GAN的基础原理淦,就是这么读。有很多种的英文字母,取名是很难的。
机器生成二次元人物的脸。
输入一个正态分布,生成一个低维的向量,会生成一个二次元人物的脸,产生一个非常高维的向量,输入不同,输出就会不同。选择一个简单的分布函数。
Discriminator输入一张图片,输出一个数字,判断一个图片是二次元人物的可能性。只是 ...
deeplearning 鱼与熊掌可以兼得
鱼与熊掌兼得(deeplearning的好处)一.两难境地在deep learning过程中,对应的参数量过多,可能会造成过拟合的现象,而相反的,参数量过少有可能获得不了比较良好的效果,这让我们进入了两难的境地。
二.piecewise linear(分段线性曲线回顾)分段线性曲线可以看作常数项加上一系列的Z形函数。
而对应的hard sigmoid的一段函数又可以有对应的两端RELU函数进行组合表示。
三.是否深度学习表现更加优异
可以看到,随着对应的深度学习的层数增加对应的语音识别的错误率也有所降低。
那么瘦高的神经网络,和矮胖的神经网络哪个更好。
若采取同样的参数量,这里两种神经网络那个更加的优秀。
如图所示,采用相同的参数量,对应的更深的神经网络的表现要明显的优于矮胖的神经网络。
甚至到最后矮胖的神经网络随着参数的增多,对应的实验效果变得不那么优秀,甚至出现了倒退的行为。
四.为什么我们需要更深的神经网络。的确一层的神经网络可以拟合出任何的函数,但是利用深度学习网络对于效果的提升更加具有效率。例如:逻辑元器件进行组合,采用更深的 组合可以减少器件的数量。在编程的过程中比 ...
CNN
CNN的主要流程①规格化图片在运行训练过程之前们需要将所有的图片scale成大小一样的对应图片,最终cross entropy越小越好,对应的误差也就越小。
模型的输入是对应的图片是一个三维矩阵,对应的前两位是对应的像素点的位置。而第三维是对应的rgb的一个3元数组,经过三维方向的拉直,形成一个可以用来训练的向量集合。
对应的机器和人都是会产生差异的,人和机器都是去寻找对应的图片中生物的特征之后就将这样的特征与人们所熟知的生物特征进行比对,形成结论,但是有可能出现错误。
例如如上的图片,无论是人还是机器都会将其识别成一个鸟类:乌鸦,但是实际上这是一只猫,一只黑猫
②分区观察自己的小范围去判断特征,这是每一个neuron所作的事情。
将对应的小范围进行拉直,作为输入,之后利用多层的运算形成最终的判断。对应的范围的属性与分布是可以根据实际情况进行灵活调整的。多个范围之间可以进行重叠,亦可以两个神经元去守备同一块的范围,非正方形的范围也是可取的。理论上对应的范围也是可以不相连的。但是实际的图像识别中很难利用的上
③最经典的设计对应的所有的channel都参与识别但是对应的小区域的范围 ...

MACHINE
Final ProjectTeam Info
English Name
Chinese Name
ID
Jing Shuji
荆树吉
202000130199
Zeng Junhao
曾俊豪
202000130222
Dataset1: Bank Marketing(classification)Assignment1Background Based on the classic marketing dataset of banks, the user characteristics and the current status of bank deposit business are analyzed to formulate bank marketing strategies. Major domestic banks and Internet wealth management institutions can learn from the marketing of bank deposit products.These data are rel ...
LD_clumping(连锁不平衡性聚类)
1.连锁不平衡性定义连锁不平衡性(linkage disequilibrium)是指 不同基因座(loci)的等位基因(allele)之间非随机(nonrandom)的关联。
两个基因座互相独立不相关,即连锁平衡 linkage equilibrium 的状态。
常用的指标:D’, r2(相关系数,correlation coefficient)【更常用】 当D’=0,r2=0时,处于完全连锁平衡状态 当D’=1,r2=1时,处于完全连锁不平衡状态。 其中,从0-1之间的度量越高,LD越高,如果两个位点连锁,连锁程度也越高。
对应的连锁不平衡性越明显,说明对应位置的相关性就越强,他们就越可能在同一种表型中发挥相似的作用。
2.LD_clumping的定义LD clumping(连锁不平衡修剪)是全基因组关联分析(GWAS)中常用的步骤,用于筛选与目标表型关联最强的独立SNP,同时移除与其高度连锁(即存在强LD)的冗余SNP。
算法原理:
索引SNP选择:从GWAS结果中筛选p值小于--clump-p1的SNP,按p值升序排序作为候选索引SN ...

PheWAS
PheWAS R语言包示例代码运行1.R环境配置(VSCODE)安装vscode(默认已经安装)安装R:安装链接https://cran.r-project.org/mirrors.html,采用国内的清华镜像站
选取第一个清华的镜像站
选取适合自己电脑的版本
下载成功
下载成功后安装的硬盘一般默认C盘,但是不要选择装在C盘,例如我就装在D盘。
2.VSCODE上安装R语言的相关插件
如果对于编辑的界面有要求可以选择安装R LSP Client插件和Radian的编辑器,(暂时安装不成功,略过)
3.测试
建立一个.R的文件,直接编译运行即可
绘图结果:
2.R环境的PheWAS的代码运行下载对应的PheWAS的安装包The PheWAS R package is designed to provide an accessible interface to the phenome wide association study.
PheWAS R软件包旨在为全表型关联研究提供一个可访问的界面。
可以使用devtools包安装PheWAS包。在R中执行以下代码将帮助您开始:
1 ...
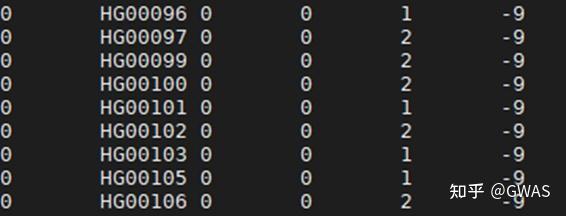
GWAS数据的介绍
样本数据集来自1000个基因组项目第3阶段第5版的504名EAS个体
-CHB:中国北京的汉族人-JPT:日本东京的日本人-CHS:南方汉族-CDX:中国西双版纳的中国傣族-KHV:金在胡志明市,越南
Url: http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20130502/
Genome build: human_g1k_v37.fasta (hg19)
基因组构建:人类_g1k_v37.fasta(hg19)
基因型数据处理
Selected only autosomal variants
Split multi-allelic variants
Variants were normalized
Remove duplicated variants
Selected only SNP (ATCG)
Selected 2% rare SNPs (plink --mac 2 --max--maf 0.01 --thin 0.02)
Selected 15% common SNPs (plink --maf 0.01 --thi ...